ODE Integrators
Available integrators
We use a high-order implicit ODE solver for integrating the reaction
system. A few alternatives, including first order implicit and explicit integrators are also
provided. Internally, the integrators use different data structures
to store the integration progress, and each integrator needs to
provide a routine to convert from the integrator’s internal
representation to the burn_t type required by the actual_rhs
and actual_jac routine.
Note
The integrator is chosen at compile-time using
the INTEGRATOR_DIR variable in the makefile.
Presently, allowed integrators are:
BackwardEuler: an implicit first-order accurate backward-Euler method. An error estimate is done by taking 2 half steps and comparing to a single full step. This error is then used to control the timestep by using the local truncation error scaling. Optionally, the user can disable error estimation and force a single-step backward-Euler integration by settingintegrator.do_single_step = 1.ForwardEuler: an explicit first-order forward-Euler method. This is meant for testing purposes only. No Jacobian is needed.QSS: the quasi-steady-state method of [42] (see also [43]). This uses a second-order predictor-corrector method, and is designed specifically for handling coupled ODE systems for chemical and nuclear reactions. However, this integrator has difficulty near NSE, so we don’t recommend its use in production for nuclear astrophysics.
RKC: a stabilized explicit Runge-Kutta-Chebyshev integrator based on [44]. This does not require a Jacobian, but does need to estimate the spectral radius of the system, which is done internally. This works for moderately stiff problems.The spectral radius is estimated by default using the power method, built into RKC. Alternately, by setting
integrator.use_circle_theorem=1, the Gershgorin circle theorem is used instead.
Rosenbrock: this implements several different Rosenbrock implicit methods for stiff ODEs. Several different methods can be selected through the runtime parameterintegrator.rosenbrock_tableau. The allowed values are:0: Rodas5P method (this is the default), based on the Julia implementation in DifferentialEquations.jl [45].1: Rodas4P method fromDifferentialEquations.jl(they give [46] as a reference).2: Rodas3P method fromDifferentialEquations.jl(see a juliacon preprint).3: ROS2S method, a 2nd order, stiff-accurate method [47].
Here the “P” suffix refers to methods developed to satisfy the stiff accuracy conditions of [48] (ROS2S also satisfies these).
The H211b error-history timestep controller from [49] (see Eq. 31) is used.
VODE: the VODE [50] integration package. We ported this integrator to C++ and removed the non-stiff integration code paths.Note
The VODE integrator uses Jacobian caching when run on a CPU by default. This can be disabled at runtime by setting
integrator.use_jacobian_caching = 0.On GPUs, we disable Jacobian caching due to the increased memory needs. Jacobian caching on GPUs can be enabled by explicitly setting the build parameter
USE_JACOBIAN_CACHING=TRUE.
We recommend that you use the VODE solver, as it is the most robust.
Note
The runtime parameter integrator.scale_system
will scale the internal energy that the integrator sees by the initial
value of \(e\) to make the system \(\mathcal{O}(1)\). The value
of atol_enuc will likewise be scaled. This works for both Strang
and simplified-SDC. For the RKC integrator, this is enabled by
default.
For most integrators this algebraic change should not affect the output to more than roundoff, but the option is included to allow for some different integration approaches in the future.
This option currently does not work with the ForwardEuler or QSS integrators.
Timestep selection
All of the integrators will select the timestep internally to meet the tolerances. There are 2 controls that affect timestepping:
integrator.ode_max_dt: sets the maximum allowed timestepintegrator.ode_max_steps: sets the maximum number of steps the integrator is allowed to take. If it exceeds this, then it will return an error.
Linear algebra
All implicit integrators use the LINPACK LU decomposition routines.
For the templated networks (aprox13, aprox19, …) the implementation
is done using consexpr loops over the equations and no pivoting is allowed.
For the other networks (usually pynucastro networks), the implementation is
provided in Microphysics/util/linpack.H and is templated on the number
of equations. Pivoting can be disabled by setting integrator.linalg_do_pivoting=0.
Tip
The storage for the Jacobian can take up the most memory when integrating the reaction system. It is possible to store the Jacobian as single-precision, by building with:
USE_SINGLE_PRECISION_JACOBIAN=TRUE
This can speed up the integration prevent the code from running out of memory when run on GPUs.
Integration errors
Important
The integrator will not abort if it encounters trouble. Instead it will
set burn_t burn_state.success = false on exit. It is up to the
application code to handle the failure.
The burn_t error_code field will provide an error code that can be
used to interpret the failure. The current codes are:
code |
meaning |
|---|---|
1 |
success |
-1 |
invalid inputs |
-2 |
underflow in computing \(\Delta t\) |
-3 |
spectral radius estimation did not converge |
-4 |
too many steps needed |
-5 |
unable to meet the accuracy demanded by the tolerances |
-6 |
non-convergence in the corrector iteration |
-7 |
LU decomposition failed |
-100 |
entered NSE |
Tolerances
Tolerances dictate how accurate the ODE solver must be while solving equations during a simulation. Typically, the smaller the tolerance is, the more accurate the results will be. However, if the tolerance is too small, the code may run for too long, the ODE solver will never converge, or it might require at timestep that underflows.
Tip
The SUNDIALS CVODE documentation on tolerances provides a good discussion on tolerances that apply here.
There are separate tolerances for the mass fractions and the energy, and there are both relative and absolute tolerances which act together. The tolerances are:
integrator.rtol_spec: the relative tolerance for the species (mass fractions when running with Strang and partial densities when running with SDC).integrator.rtol_enuc: the relative tolerance on the energy (specific internal energy when running with Strang, internal energy density when running with SDC).integrator.atol_spec: the absolute tolerance for the species (this is always interpreted in terms of mass fraction and the appropriate density weighting will be added for SDC).integrator.atol_enuc: the absolute tolerance for energy – this is generally not interesting, since the energy is so large and therefore best served via a relative tolerance.
The tolerances are combined, e.g. for species, as:
so if the mass fraction, \(X_k\), is very small, then the absolute tolerance will set the error that the integrator tries to achieve. If the mass fraction is large, \(\mathcal{O}(1)\), then the relative tolerance dominates.
Some suggestions when setting tolerances:
If a burn does not converge with one type of Jacobian (analytic or numerical) then it may do better with the other type. This can be automated via the
integrator.use_burn_retrymechanism described above.Sometimes a burn completes better if the absolute tolerances are made even smaller – this will require the integrator to track trace species better which can help with equilibrium better.
The VODE integrator has additional logic meant to ensure that species don’t change too much per timestep. This is controlled by
integrator.X_reject_buffer. If a species \(k\), has a mass fraction \(X_k > \mbox{X\_reject\_buffer} \cdot \mbox{atol\_spec}\) then we reject a VODE timestep if the mass fraction changes by more than a factor of 4 in a single VODE timestep and we try again. This is all done internally to VODE. MakingX_reject_bufferlarger will allow it to ignore more trace species.
Below is a comparison of how the tolerances affect the nucleosynthesis.
This is run using burn_cell and the aprox13 network. Four separate
runs were done, using tolerances of \(10^{-3}\), \(10^{-5}\), \(10^{-8}\), and \(10^{-12}\)
(all 4 tolerance parameters were set to the same value). The run with the tightest
tolerances (\(10^{-12}\)) was taken as the reference and relative errors were
computed with respect to it. The scripts for this are in Microphysics/unit_test/burn_cell/compare_tolerances/.
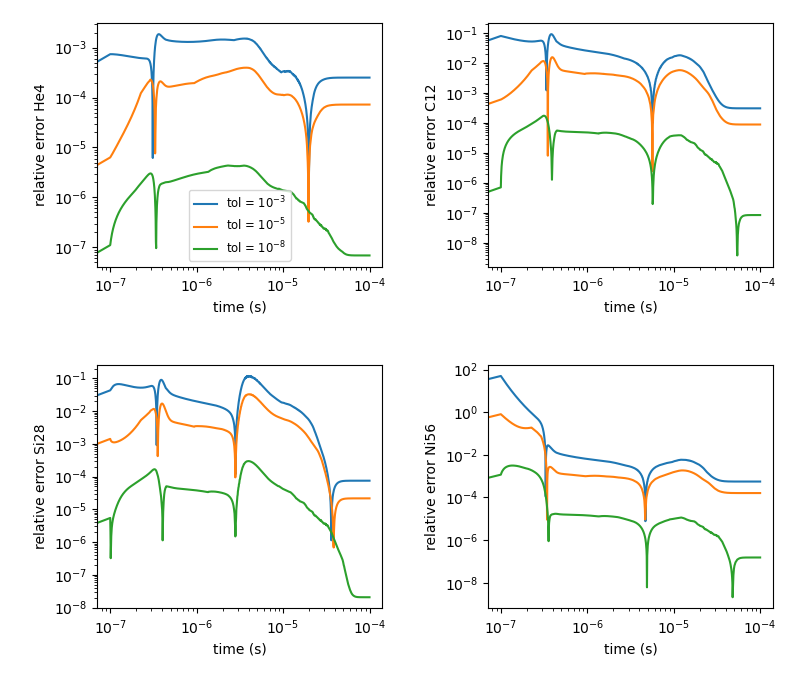
Fig. 1 Relative error of runs with varying tolerances as compared to a run with an ODE tolerance of \(10^{-12}\).
We see that using a tolerance of \(10^{-5}\) generally gives reasonable mass fractions. Looser than this can produce large errors.
Controlling Species \(\sum_k X_k = 1\)
The ODE integrators don’t know about the constraint that
so this is only going to be preserved to the level that the integrator tolerances allow. There are a few parameters that help enforce this constraint on the intermediate states during the integration.
integrator.renormalize_abundances: this controls whether we renormalize the abundances so that the mass fractions sum to one during a burn.This has the positive benefit that in some cases it can prevent the integrator from going off to infinity or otherwise go crazy; a possible negative benefit is that it may slow down convergence because it interferes with the integration scheme. Regardless of whether you enable this, we will always ensure that the mass fractions stay positive and larger than some floor
small_x.This option is disabled by default.
integrator.SMALL_X_SAFE: this is the floor on the mass fractions. The default is1.e-30.integrator.do_species_clip: this enforces that the mass fractions all in \([\mathtt{SMALL\_X\_SAFE}, 1.0]\) before calling the network righthand side function.This is off by default. Turning this on can sometimes make the integrator work a lot harder.
integrator.do_corrector_validation: in the nonlinear solve corrector loop, when we get a corrected integration state, do we check to make sure the mass fractions are valid before calling the righthand function? This is needed in some cases ifintegrator.do_species_clipis disabled. Note: this is not implemented for every integrator.
Retry Mechanism
Integration can fail for a number of reasons. Some of the errors you may see are:
Not enough steps allowed (
integrator.ode_max_steps)The timestep selected by the integrator is too small (comparable to roundoff)
The final abundances do not sum to 1.
There can be a number of reasons for these failures, including:
The Jacobian is not accurate enough
This can lead to issues 1 or 2 above
The integrator is not appropriate for the thermodynamic conditions
For example, the RKC integrator may be working too hard, leading to issue 1.
The tolerances you are requesting are too tight
This can lead to issues 1 or 2 above
The tolerances (in particular,
integrator.atol_spec) are too looseThis can lead to issue 3 above
The evolution is entering NSE
This can lead to issue 1.
The integrator() function that calls the actual integrator drive for
the choice of integrator allows for a retry if a burn failure was detected.
This is enabled by setting
integrator.use_burn_retry = 1
This will call the same integrator again, restarting from the initial conditions but with a different choice of tolerances and Jacobian. The runtime parameters that come into play when doing the retry are:
retry_swap_jacobian: do we swap that Jacobian type for the retry (i.e. use the numerical Jacobian if we try the analytic Jacobian for the first attempt)retry_rtol_spec: relative tolerance for the species on retryretry_rtol_enuc: relative tolerance for the energy on retryretry_atol_spec: absolute tolerance for the species on retryretry_atol_enuc: absolute tolerance for the energy on retry
Note
If you set any of the retry tolerances to be less than \(0\), then the original (non-retry) tolerance is used on retry. The default value for all of the retry tolerances is \(-1\), which means the same tolerances are used on retry unless you override them at runtime.
Tip
Sometimes a simulation runs best if you set
integrator.ode_max_steps to a small value (like 10000) and
start with the analytic Jacobian (integrator.jacobian = 1) and
then use the retry mechanism to swap the Jacobian on any zones that fail.