burn_cell
burn_cell is a simple one-zone burn that will evolve a state with
a network for a specified amount of time. This can be used to
understand the timescales involved in a reaction sequence or to
determine the needed ODE tolerances. This is designed to work
with the Strang-split integration wrappers. The system that is evolved
has the form:
with density held constant and the temperature found via the equation of state, \(T = T(\rho, X_k, e)\).
Note
Since the energy evolves due to the heat release (or loss)
from reactions, the temperature will change during the burn
(unless integrator.call_eos_in_rhs=0 is set).
Getting Started
The burn_cell code is located in
Microphysics/unit_test/burn_cell. An inputs file which sets the
default parameters for your choice of network is needed to run the
test. There are a number of inputs files in the unit test directory
already with a name list inputs_network, where network
is the network you wish to use for your testing. These can be
used as a starting point for any explorations.
Setting the thermodynamics
The parameters that affect the thermodynamics are:
unit_test.density: the initial densityunit_test.temperature: the initial temperature
The composition can be set either by specifying individual mass fractions
or setting unit_test.uniform_xn as described in Defining composition.
If the values don’t sum to 1 initially, then the test will do a
normalization. This normalization can be disabled by setting:
unit_test.skip_initial_normalization = 1
Controlling time
The test will run unit a time unit_test.tmax, outputting the state
at regular intervals. The parameters controlling the output are:
unit_test.tmax: the end point of integration.unit_test.tfirst: the first time interval to output.unit_test.nsteps: the number of steps to divide the integration into, logarithmically-spaced.
If there is only a single step, unit_test.nsteps = 1, then we integrate
from \([0, \mathrm{tmax}]\).
If there are multiple steps, then the first output will be at a time \(\mathrm{tmax} / \mathrm{nsteps}\), and the steps will be logarithmically-spaced afterwards.
Integration parameters
The tolerances, choice of Jacobian, and other integration parameters
can be set via the usual Microphysics runtime parameters, e.g.
integrator.atol_spec.
Building and Running the Code
The code can be built simply as:
make
and the network and integrator can be changed using the normal Microphysics build system parameters, e.g.,
make NETWORK_DIR=aprox19 INTEGRATOR_DIR=rkc
The build process will automatically create links in the build directory to the EOS table and any reaction rate tables needed by your choice of network.
Important
You need to do a make clean before rebuilding with a different
network or integrator.
To run the code, in the burn_cell directory run:
./main3d.gnu.ex inputs
where inputs is the name of your inputs file.
Working with Output
Note
For this part, we’ll assume that the default aprox13 and
VODE options were used for the network and integrator, and the
test was run with inputs.aprox13.
As the code runs, it will output to stdout details of the initial
and final state and the number of integration steps taken (along with whether
the burn was successful). The full history of the thermodynamic state will also be output to a file,
state_over_time.txt, with each line corresponding to one of the
nsteps requested in the time integration.
The script plot_burn_cell.py can be used to visualize the evolution:
python plot_burn_cell.py state_over_time.txt
This will generate the following figure:
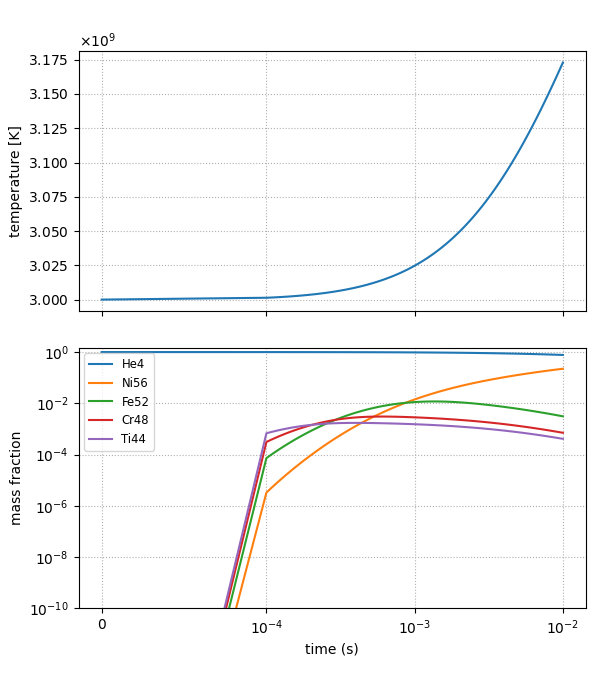
Only the most abundant species are plotted. The number of species to plot and the
limits of \(X\) can be set via runtime parameters (see python plot_burn_cell.py -h).